.
Algébriquement (en notant la masse volumique, uniforme dans ce cas) :
.
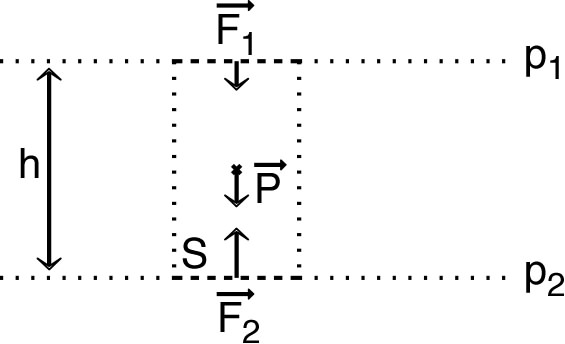
Les forces latérales se compensent par
symétrie ; donc à l’équilibre :. Algébriquement (en notant la masse volumique, uniforme dans ce cas) : . |
|
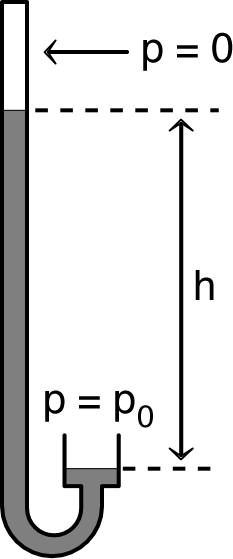
| • La loi d’évolution de la pression selon
la profondeur est le principe de base des manomètres à
dénivellation de liquide. Sous la pression normale, la dénivellation dans un baromètre à mercure est : (avec à la température usuelle). 📖 exercices n° II, III, IV et V. |
 |
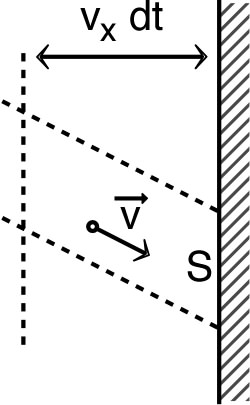
| Ainsi, la force pressante d'un gaz sur une
surface
correspond à la quantité de mouvement
transférée pendant un temps
:
. Ceci est indépendant des autres composantes du mouvement ; on peut donc raisonner comme si tous les constituants se déplaçaient selon . |
 |
;et la “vitesse quadratique moyenne” est telle que :
;
.
(donc à la concentration des constituants) ;
.
;
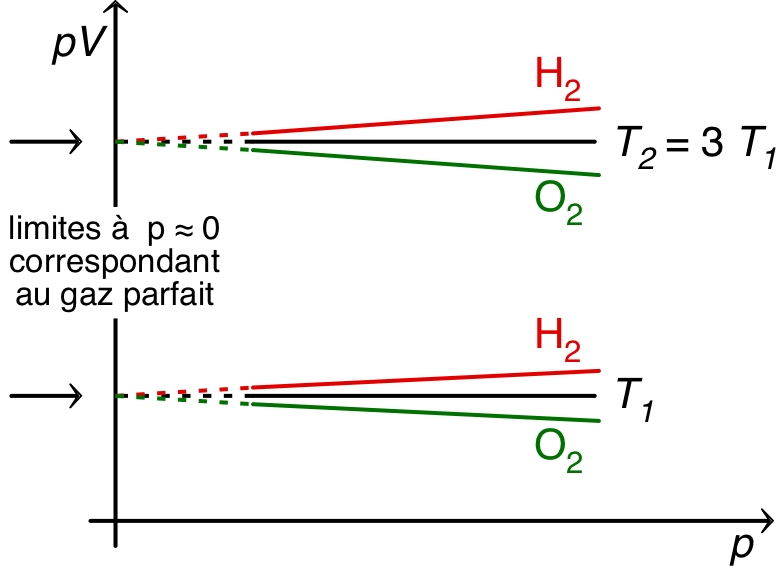
| • Le diagramme d’Amagat (tracé pour
fixé)
montre bien l’approximation du modèle des gaz parfaits. ◊ remarque : on sait d'ailleurs qu'aux températures usuelles, le gaz se liquéfie si on augmente suffisamment la pression. |
 |
.
dilatation isobare ;
compressibilité isotherme.
; .