MESURE D’UNE ENTHALPIE DE RÉACTION - corrigé du TP
Enthalpie de fusion de la glace et méthode des mélanges
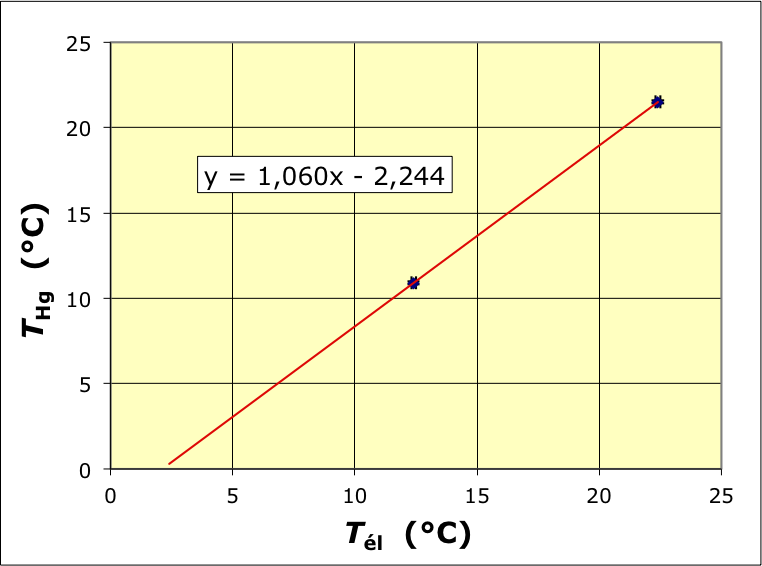
• Les étudiants du groupe de TP disposaient d'un thermomètre électronique, assurément capable de précision et plus facile à manipuler, mais doutaient un peu de ses réglages dans le processus de fabrication à la chaîne.
Ils ont procédé à un étalonnage en deux points (dans la zone des températures concernées) par comparaison à un thermomètre à mercure dont le fonctionnement avait précédemment déjà été vérifié à de nombreuses reprises.
On constate que des écarts peuvent atteindre
6
%
6 %
; les valeurs lues sur l'affichage sont par suite corrigées en conséquence.
• La manipulation est réalisée à la température extérieure
T
e
=
21
,
0
±
0
,
1
°
C
T_e=21\text{,}0±0\text{,}1 \:\mathrm{°C}
.
La cuve d'aluminium du calorimètre et l'agitateur ont une masse
m
0
=
99
,
3
±
0
,
2
g
m_0=99\text{,}3±0\text{,}2\:\mathrm{g}
; avec une capacité calorifique massique (lue dans les tables)
c
0
=
0
,
89
±
0
,
01
J
.
g
−
1
.
K
−
1
c_0=0\text{,}89±0\text{,}01 \;\mathrm{J .g^{-1}.K^{-1}}
(précision modeste car des alliages sont généralement utilisés pour rendre le métal plus rigide) cela correspond à
C
0
=
88
,
4
±
1
,
2
J
.
K
−
1
C_0=88\text{,}4±1\text{,}2 \;\mathrm{J .K^{-1}}
.
La masse d'eau initiale est
m
1
=
233
,
6
±
0
,
2
g
m_1=233\text{,}6±0\text{,}2\:\mathrm{g}
est à une température initiale
T
1
=
12
,
5
±
0
,
1
°
C
T_1=12\text{,}5±0\text{,}1 \:\mathrm{°C}
(on peut estimer que la cuve du calorimètre aussi, en approximation raisonnable) ; avec une capacité calorifique massique
c
1
=
4
,
18
±
0
,
01
J
.
g
−
1
.
K
−
1
c_1=4\text{,}18±0\text{,}01 \;\mathrm{J .g^{-1}.K^{-1}}
cela correspond à
C
1
=
88
,
4
±
1
,
2
J
.
K
−
1
C_1=88\text{,}4±1\text{,}2 \;\mathrm{J .K^{-1}}
.
La masse du glaçon est
m
2
=
42
,
9
±
0
,
2
g
m_2=42\text{,}9±0\text{,}2\:\mathrm{g}
; la capacité calorifique massique
c
2
=
2
,
10
±
0
,
01
J
.
g
−
1
.
K
−
1
c_2=2\text{,}10±0\text{,}01 \;\mathrm{J .g^{-1}.K^{-1}}
de la glace n'intervient pas puisque le glaçon est initialement à
0
°
C
0 \:\mathrm{°C}
; pour l'eau de fusion de la glace cela correspond à
C
2
=
179
,
3
±
1
,
3
J
.
K
−
1
C_2=179\text{,}3±1\text{,}3 \;\mathrm{J .K^{-1}}
.
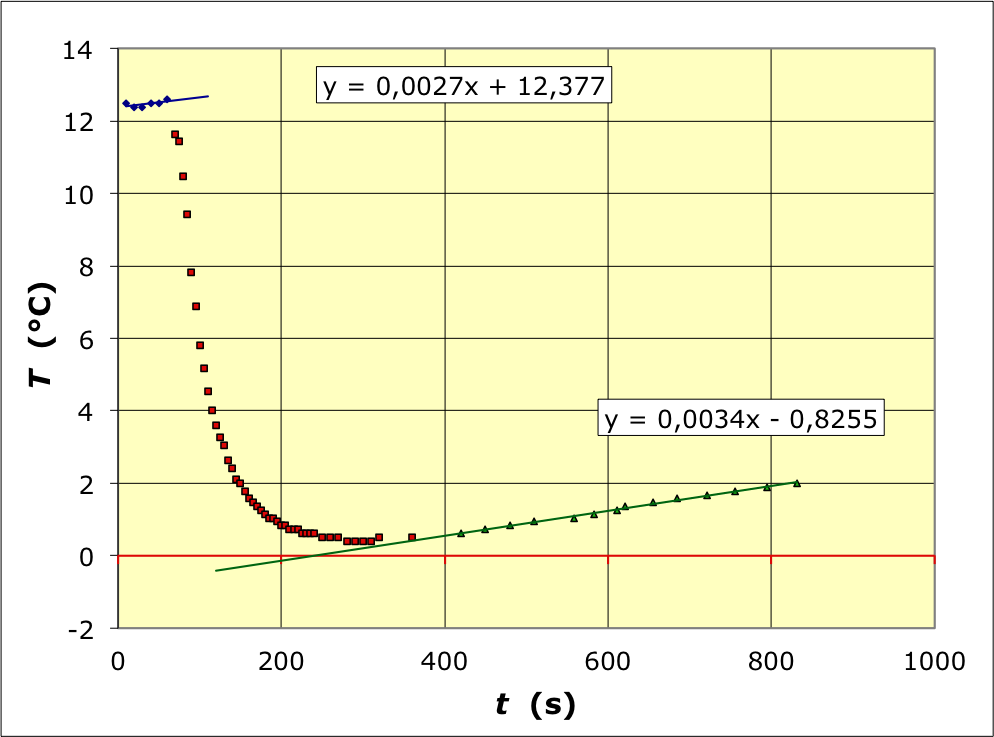
• On mesure la température avant, pendant et après la fusion :
• En procédant de façon la plus simpliste, on repère la température minimale
T
=
0
,
4
±
0
,
2
°
C
T=0\text{,}4±0\text{,}2 \:\mathrm{°C}
. Cela correspond à une variation
∆
T
01
=
−
12
,
1
±
0
,
3
°
C
∆T_{01}=-12\text{,}1±0\text{,}3 \:\mathrm{°C}
.
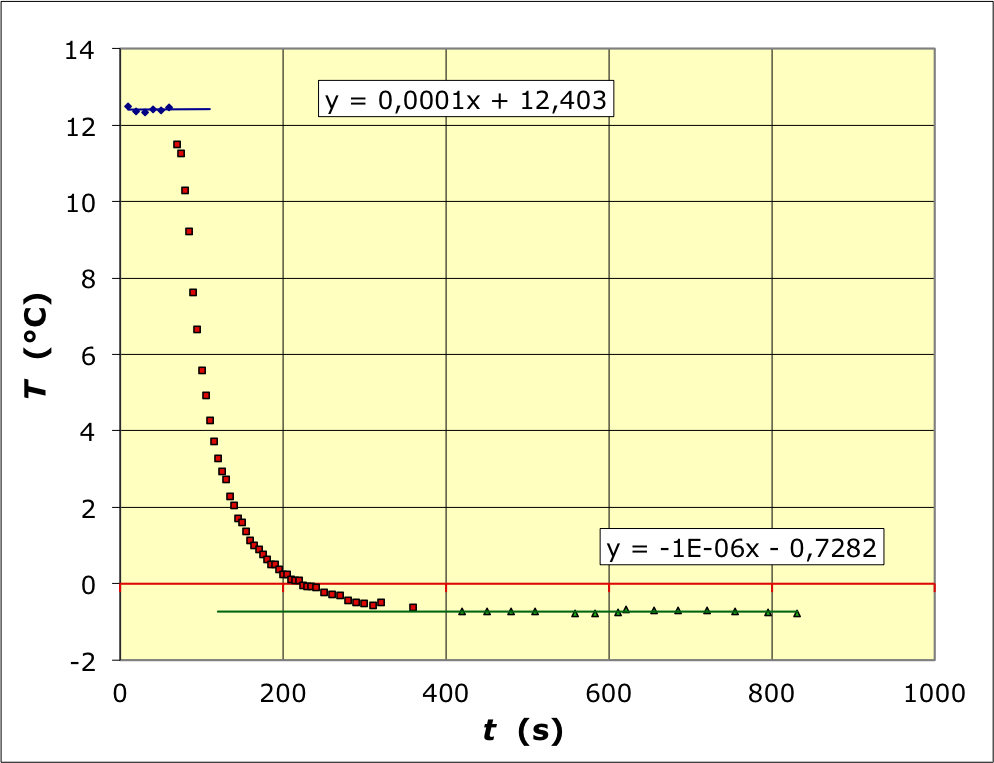
Les ajustements affines sur les portions avant et après fusion peuvent être prolongés jusqu'aux environs du milieu de la réaction. Puisque la variation
T
(
t
)
T(t)
pendant cette dernière n'est pas affine, on peut se demander jusqu'à quel instant il est préférable de prolonger ; la disposition des courbes est toutefois telle (les droites avant et après sont presque parallèles) que cela importe peu ; l'effet est très nettement du second ordre. On obtient ainsi la valeur corrigée
∆
T
01
=
−
13
,
1
±
0
,
3
°
C
∆T_{01}=-13\text{,}1±0\text{,}3 \:\mathrm{°C}
.
Cela donne :
𝓁
p
=
−
(
C
0
+
C
1
)
∆
T
01
+
C
2
∆
T
2
m
2
=
329
±
10
J
.
g
−
1
\displaystyle 𝓁_p=-\frac{(C_0+C_1 ) \: ∆T_{01}+C_2 \: ∆T_2}{m_2} =329±10 \;\mathrm{J .g^{-1}}
bien compatible avec la valeur
𝓁
p
=
335
±
1
J
.
g
−
1
𝓁_p=335±1 \;\mathrm{J .g^{-1}}
généralement admise par la communauté scientifique.
• Une autre présentation est possible en compensant, en fonction de
t
t
, le flux thermique parasite décrit par l'expression affine
d
Q
d
t
=
A
+
B
.
(
T
−
T
e
)
\displaystyle \frac{dQ}{dt}=A+B.(T-T_e)
et en ajustant les constantes
A
A
et
B
B
de façon à rendre les portions avant et après horizontales.
Le résultat est le même (la différence des deux méthodes est du second ordre) ; il s'agit simplement d'une autre présentation, dont l'avantage est de mieux montrer l'efficacité de la correction, mais qui a l'inconvénient de masquer un peu la démarche dans les calculs délégués au logiciel.
Enthalpie de vaporisation de l’eau et chauffage électrique
• On peut éventuellement s'intéresser ici à préciser certains détails de la modélisation théorique.
• Avec l’énergie interne
U
U
, on peut écrire en général :
d
U
=
δ
Q
+
δ
W
n
c
dU=δQ+δW_{nc}
où
δ
W
n
c
=
−
p
d
V
+
𝒰
d
q
δW_{nc}=-p \:dV+𝒰 \:dq
représente le travail des forces “non conservatives” (c’est-à-dire non prises en compte par un terme d’énergie potentielle dans l’énergie mécanique), c’est-à-dire ici les forces pressantes et les forces électriques microscopiques dont la conséquence est l’effet Joule.
Avec l’enthalpie, ceci s’écrit :
d
H
=
d
(
U
+
p
V
)
=
V
d
p
+
δ
Q
+
δ
W
n
c
n
p
dH=d(U+p \:V)=V \:dp+δQ+δW_{ncnp}
où
δ
W
n
c
n
p
=
𝒰
d
q
=
𝒰
I
d
t
δW_{ncnp}=𝒰 \:dq=𝒰 \:I \:dt
est le travail des forces “non conservatives” autres que les forces pressantes.
Le système étant thermiquement “isolé” (
δ
Q
=
0
δQ=0
) à pression constante (
d
p
=
0
dp=0
), il reste :
d
H
=
δ
W
n
c
n
p
dH=δW_{ncnp}
et par intégration :
∆
H
=
𝒰
I
∆
t
∆H=𝒰 \:I \:∆t
si on manipule à puissance électrique
P
=
𝒰
I
P=𝒰 \:I
constante.
• Mais par ailleurs, puisque le système ne contient aucune partie du type pile ou condensateur (capable d’accumuler l’effet de la charge qui circule),
H
H
ne peut pas dépendre de
q
q
(ce n’est pas, dans ce cas, une variable d’état) :
H
=
H
(
T
,
p
,
m
)
H=H(T,p,m)
.
On peut donc écrire :
d
H
=
[
∂
H
∂
T
]
p
,
m
d
T
+
[
∂
H
∂
p
]
T
,
m
d
p
+
[
∂
H
∂
m
]
T
,
p
d
m
=
𝓁
p
d
m
\displaystyle dH=\left[\frac{∂H}{∂T}\right]_{p,m} \: dT+\left[\frac{∂H}{∂p}\right]_{T,m} \: dp+\left[\frac{∂H}{∂m}\right]_{T,p} \: dm=𝓁_p \: dm
quand on manipule à pression constante (
d
p
=
0
dp=0
) et en régime stationnaire à la température d’ébullition (
d
T
=
0
dT=0
).
On obtient ainsi :
∆
H
=
𝓁
p
m
=
𝒰
I
∆
t
∆H=𝓁_p \: m=𝒰 \:I \:∆t
et on peut donc en déduire :
𝓁
p
=
𝒰
I
∆
t
m
\displaystyle 𝓁_p=𝒰 \:I \, \frac{∆t}{m}
.
• En s'organisant à plusieurs groupes (ceux travaillant aux plus faibles puissances n'ont le temps de faire qu'une mesure), les étudiants ont effectué sept mesures pour des puissances électriques différentes :
n° mesure
1
2
3
4
5
6
7
I
(
A
)
I \;(\mathrm{A})
4
,
00
±
0
,
04
4\text{,}00±0\text{,}04
3
,
02
±
0
,
04
3\text{,}02±0\text{,}04
3
,
02
±
0
,
04
3\text{,}02±0\text{,}04
2
,
40
±
0
,
03
2\text{,}40±0\text{,}03
3
,
33
±
0
,
04
3\text{,}33±0\text{,}04
1
,
54
±
0
,
03
1\text{,}54±0\text{,}03
4
,
00
±
0
,
04
4\text{,}00±0\text{,}04
𝒰
(
V
)
𝒰 \;(\mathrm{V})
19
,
2
±
0
,
3
19\text{,}2±0\text{,}3
14
,
5
±
0
,
3
14\text{,}5±0\text{,}3
14
,
4
±
0
,
3
14\text{,}4±0\text{,}3
11
,
6
±
0
,
3
11\text{,}6±0\text{,}3
16
,
1
±
0
,
3
16\text{,}1±0\text{,}3
7
,
4
±
0
,
2
7\text{,}4±0\text{,}2
19
,
3
±
0
,
3
19\text{,}3±0\text{,}3
∆
t
(
s
)
∆t \;(\mathrm{s})
540
±
5
540±5
830
±
5
830±5
720
±
5
720±5
1020
±
5
1020±5
630
±
5
630±5
1799
±
5
1799±5
780
±
5
780±5
m
(
g
)
m \;(\mathrm{g})
17
,
2
±
0
,
2
17\text{,}2±0\text{,}2
13
,
5
±
0
,
2
13\text{,}5±0\text{,}2
11
,
7
±
0
,
2
11\text{,}7±0\text{,}2
10
,
4
±
0
,
2
10\text{,}4±0\text{,}2
13
,
5
±
0
,
2
13\text{,}5±0\text{,}2
5
,
1
±
0
,
2
5\text{,}1±0\text{,}2
24
,
6
±
0
,
2
24\text{,}6±0\text{,}2
P
=
𝒰
I
(
W
)
P=𝒰 \:I \;(\mathrm{W})
76
,
8
±
2
,
0
76\text{,}8±2\text{,}0
43
,
8
±
1
,
5
43\text{,}8±1\text{,}5
43
,
5
±
1
,
5
43\text{,}5±1\text{,}5
27
,
8
±
1
,
1
27\text{,}8±1\text{,}1
53
,
6
±
1
,
6
53\text{,}6±1\text{,}6
11
,
4
±
0
,
5
11\text{,}4±0\text{,}5
77
,
2
±
2
,
0
77\text{,}2±2\text{,}0
𝓁
p
(
k
J
.
g
−
1
)
𝓁_p \; (\mathrm{kJ .g^{-1}})
2
,
41
±
0
,
11
2\text{,}41±0\text{,}11
2
,
69
±
0
,
15
2\text{,}69±0\text{,}15
2
,
68
±
0
,
16
2\text{,}68±0\text{,}16
2
,
73
±
0
,
17
2\text{,}73±0\text{,}17
2
,
50
±
0
,
13
2\text{,}50±0\text{,}13
4
,
02
±
0
,
36
4\text{,}02±0\text{,}36
2
,
45
±
0
,
10
2\text{,}45±0\text{,}10
• Même si on élimine la mesure n° 6, les autres résultats ne sont compatibles que très grossièrement. Plus précisément , cela donne l’ordre de grandeur de l’enthalpie de réaction cherchée, mais l’ajustement de la loi :
𝓁
p
=
𝒰
I
∆
t
m
\displaystyle 𝓁_p=𝒰 \:I \, \frac{∆t}{m}
(avec les incertitudes calculées) conduit à :
𝓁
p
≈
2
,
59
±
0
,
04
k
J
.
g
−
1
𝓁_p≈2\text{,}59±0\text{,}04 \;\mathrm{kJ .g^{-1}}
qui est autant incompatible avec les différentes mesures individuelles qu’avec la valeur usuellement admise par la communauté scientifique :
𝓁
p
=
2
,
256
±
0
,
014
k
J
.
g
−
1
𝓁_p=2\text{,}256±0\text{,}014 \;\mathrm{kJ .g^{-1}}
.
On constate par ailleurs que les mesures sont d’autant plus éloignées de la valeur usuellement admise qu’elles correspondent à une grande durée de manipulation, à cause d’une faible puissance électrique et donc logiquement à des fuites de chaleur proportionnellement plus grandes ; ceci conduit donc à remettre en cause la validité de l'interprétation simpliste : il faut tenir compte des “ fuites”.
• Compte tenu de la température constante (
100
°
C
100 \:\mathrm{°C}
) on peut considérer que les transferts de chaleur parasites (ici des pertes de chaleur) correspondent à une puissance
P
0
P_0
constante.
On peut en outre envisager une perte de vapeur, sortant du dispositif sans se condenser, ou se réévaporant après condensation. Les pertes de ce type sont en première approximation constantes (bien qu’elles puissent augmenter quand le débit de vapeur est plus grand) et on peut donc (pour le raisonnement formel) les inclure dans la “puissance perdue”
P
0
P_0
.
• L’équation calorimétrique donne alors :
δ
Q
=
−
P
0
d
t
δQ=-P_0 \: dt
(pertes) et conduit ainsi à :
P
=
P
0
+
𝓁
p
m
∆
t
\displaystyle P=P_0+𝓁_p \, \frac{m}{∆t}
.
On reprend alors les données pour considérer
P
P
en fonction de
m
∆
t
\displaystyle \frac{m}{∆t}
:
n° mesure
1
2
3
4
5
6
7
P
=
𝒰
I
(
W
)
P=𝒰 \:I \;(\mathrm{W})
76
,
8
±
2
,
0
76\text{,}8±2\text{,}0
43
,
8
±
1
,
5
43\text{,}8±1\text{,}5
43
,
5
±
1
,
5
43\text{,}5±1\text{,}5
27
,
8
±
1
,
1
27\text{,}8±1\text{,}1
53
,
6
±
1
,
6
53\text{,}6±1\text{,}6
11
,
4
±
0
,
5
11\text{,}4±0\text{,}5
77
,
2
±
2
,
0
77\text{,}2±2\text{,}0
m
∆
t
(
m
g
.
s
−
1
)
\displaystyle \frac{m}{∆t} \; (\mathrm{mg .s^{-1}})
31
,
9
±
0
,
7
31\text{,}9±0\text{,}7
16
,
3
±
0
,
3
16\text{,}3±0\text{,}3
16
,
3
±
0
,
4
16\text{,}3±0\text{,}4
10
,
2
±
0
,
3
10\text{,}2±0\text{,}3
21
,
4
±
0
,
4
21\text{,}4±0\text{,}4
2
,
83
±
0
,
12
2\text{,}83±0\text{,}12
31
,
5
±
0
,
5
31\text{,}5±0\text{,}5
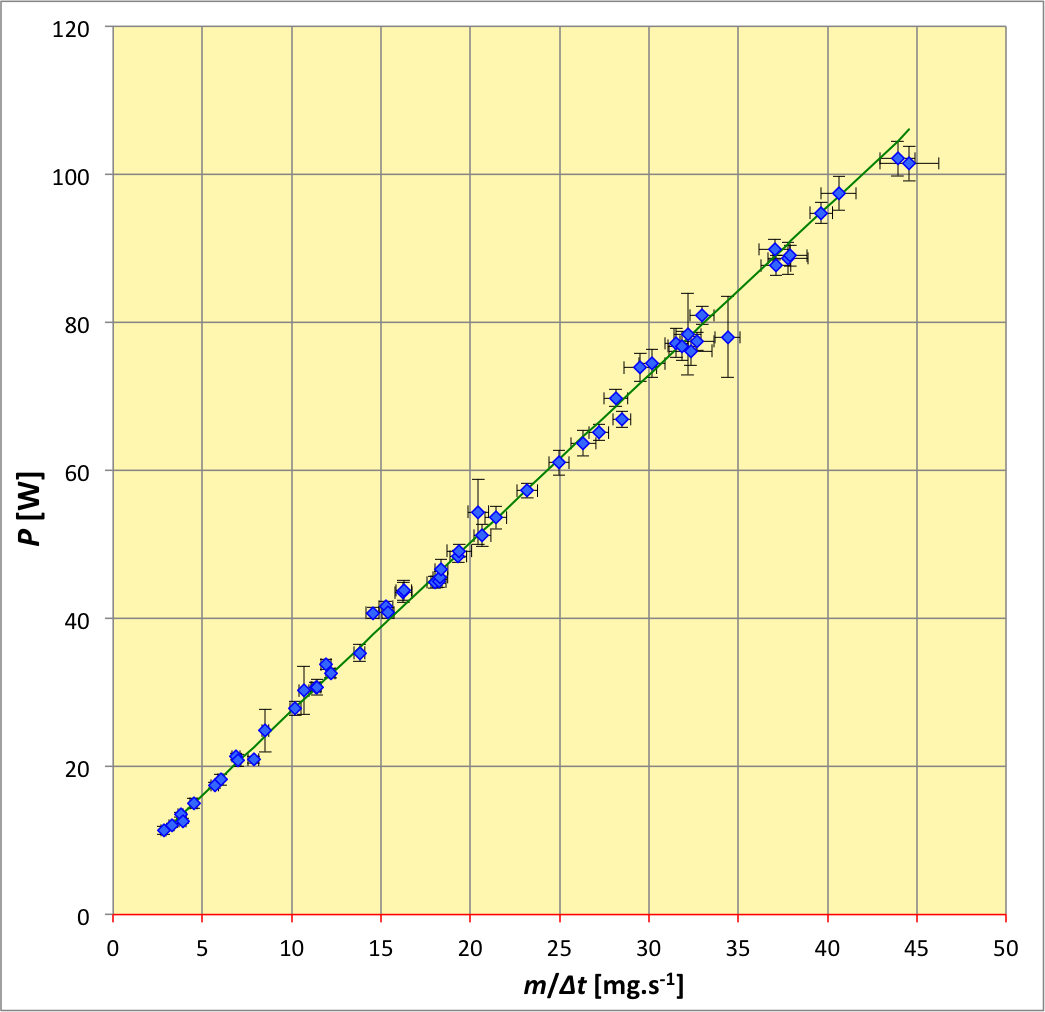
L’ajustement d’une variation affine montre que les points expérimentaux sont extrêmement bien alignés (compte tenu des incertitudes de mesure relativement faibles). On obtient ainsi la puissance perdue comme ordonnée à l’origine :
P
0
=
4
,
9
±
0
,
7
W
P_0=4\text{,}9±0\text{,}7\:\mathrm{W}
(de loin non négligeable pour la mesure n° 6) et l’enthalpie massique de réaction comme pente de la droite :
𝓁
p
=
2
,
30
±
0
,
06
k
J
.
g
−
1
𝓁_p=2\text{,}30±0\text{,}06 \;\mathrm{kJ .g^{-1}}
.
Cette détermination de l’enthalpie de réaction est non seulement compatible avec la valeur usuellement admise, mais elle est de plus d’une précision remarquable pour une expérience de calorimétrie en TP (incertitude relative
≤
3
%
≤3 \:%
).
• Il a alors été décidé de cumuler les résultats sur plusieurs années ; l'ajustement par le
χ
2
χ^2
d'une série de 55 mesures donne :
P
0
=
4
,
79
±
0
,
24
W
P_0=4\text{,}79±0\text{,}24\:\mathrm{W}
et
𝓁
p
=
2
,
271
±
0
,
018
k
J
.
g
−
1
𝓁_p=2\text{,}271±0\text{,}018 \;\mathrm{kJ .g^{-1}}
; la précision est presque aussi bonne que celle des laboratoires spécialisés.
Dosage calorimétrique et enthalpie molaire de l’autoprotolyse
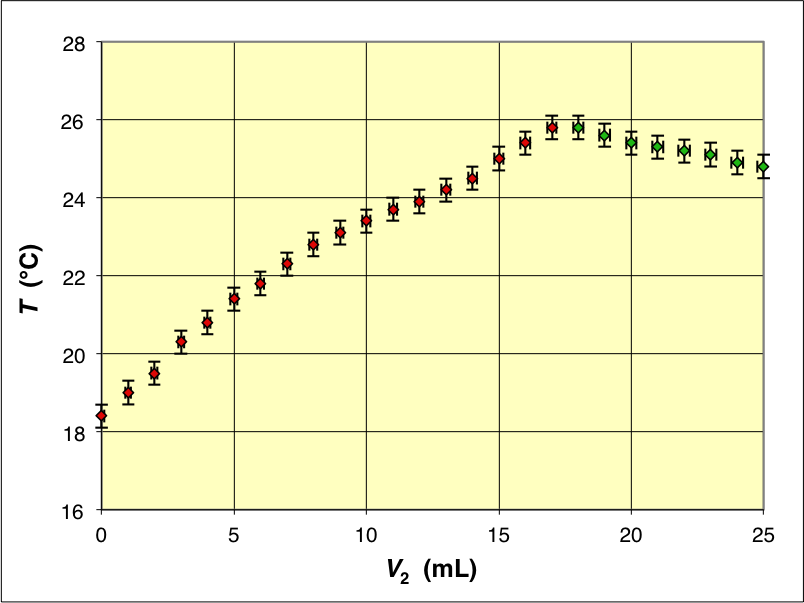
• La manipulation est réalisée à la température extérieure
T
e
=
18
,
4
±
0
,
3
°
C
T_e=18\text{,}4±0\text{,}3 \:\mathrm{°C}
. Avant la réaction, la température du calorimètre et de son contenu est stabilisée à cette valeur.
Le volume de la solution acide initiale est
V
1
=
50
,
0
±
0
,
2
m
L
V_1=50\text{,}0±0\text{,}2 \:\mathrm{mL}
; sa concentration
𝒞
a
=
1
,
00
±
0
,
02
m
o
l
.
L
−
1
𝒞_a=1\text{,}00±0\text{,}02 \;\mathrm{mol .L^{-1}}
.
◊ remarque : la fiole jaugée de
50
m
L
50 \:\mathrm{mL}
n'est pas un récipient prévu pour mesurer un volume à transférer, mais c'est plus simple et de précision acceptable dans ce cas.
• Les mesures indiquent une équivalence pour
V
e
=
17
,
3
±
0
,
3
m
L
V_e=17\text{,}3±0\text{,}3 \:\mathrm{mL}
; ceci correspond à une concentration
𝒞
b
=
2
,
89
±
0
,
06
m
o
l
.
L
−
1
𝒞_b=2\text{,}89±0\text{,}06 \;\mathrm{mol .L^{-1}}
non incompatible avec la solution préparée (
3
,
0
m
o
l
.
L
−
1
3\text{,}0 \;\mathrm{mol .L^{-1}}
) mais un peu inférieure, ce qui suggère un peu de réaction parasite de la soude avec le
C
O
2
\mathrm{CO}_2
de l'air.
◊ remarque : dans la solution de soude, le
C
O
2
\mathrm{CO}_2
se retrouve sous forme
C
O
3
2
−
\mathrm{CO}_3^{2-}
au début spectateur (moins réactif que
H
O
−
\mathrm{HO}^-
) ; mais à l'approche de l'équivalence le
p
H
\mathrm{pH}
dans la cuve du calorimètre dépasse
p
K
a
=
6
,
3
\mathrm{p}K_\mathrm{a}=6\text{,}3
et la réaction étudiée est un peu perturbée, ce qui conduit à une légère déformation de la courbe (de l'ordre de grandeur des incertitudes, mais systématique).
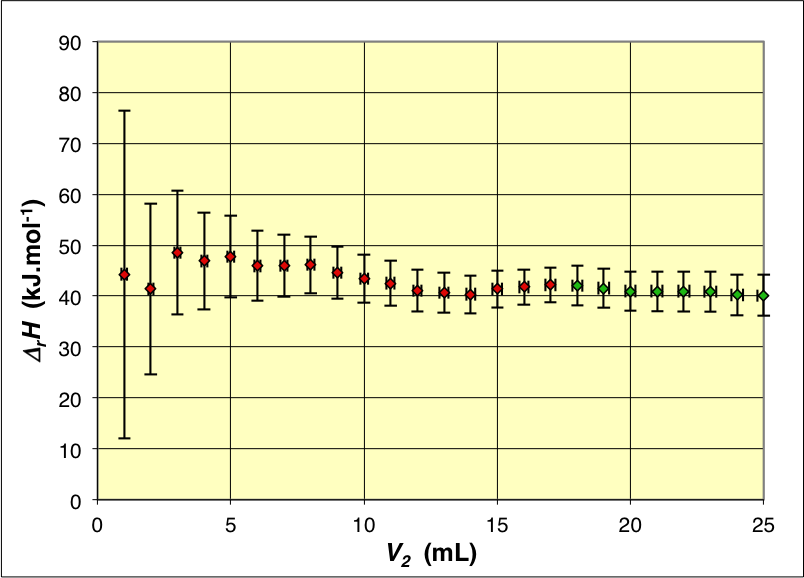
• Avant l'équivalence, la conservation de l'enthalpie donne :
∆
r
H
=
μ
c
.
(
V
1
+
V
2
)
.
(
T
0
−
T
)
𝒞
b
V
2
\displaystyle ∆_r H=\frac{μ \:c.(V_1+V_2 ).(T_0-T)}{𝒞_b \: V_2}
; de même après l'équivalence :
∆
r
H
=
μ
c
.
(
V
1
+
V
2
)
.
(
T
0
−
T
)
𝒞
b
V
e
\displaystyle ∆_r H=\frac{μ \:c.(V_1+V_2 ).(T_0-T)}{𝒞_b \: V_e}
.
◊ remarque : la cuve est un vase Dewar et sa capacité thermique est négligeable ; les solutions ont (comme l'eau) une capacité thermique massique
c
=
4
,
18
J
.
g
−
1
.
K
−
1
c=4\text{,}18 \;\mathrm{J .g^{-1}.K^{-1}}
.
• Les valeurs ainsi calculées pour les différents volumes sont relativement compatibles et on pourrait en considérer une moyenne (pondérée par les incertitudes, ou par ajustement de la courbe théorique sur les variations de température) mais on peut remarquer une légère tendance à la diminution, ce qui suggère de légères fuites thermiques.
En fait, les mesures de température après la fin de l'ajout de solution montrent une baisse de température de
≈
0
,
3
°
C
≈0,3 \:\mathrm{°C}
en deux minutes, de l'ordre de grandeur des incertitudes mais avec une tendance systématique.
• Compte tenu de la capacité thermique pour
V
2
f
=
25
m
L
V_{2f}=25 \:\mathrm{mL}
et de la température “finale”
T
f
=
24
,
8
°
C
T_f=24\text{,}8 \:\mathrm{°C}
cela correspond à une puissance de fuites
P
=
0
,
72
W
P=0\text{,}72 \:\mathrm{W}
et on peut modéliser ces pertes par un flux de la forme
d
Q
d
t
=
B
.
(
T
−
T
e
)
\displaystyle \frac{dQ}{dt}=B.(T-T_e)
avec
B
≈
0
,
116
W
.
K
−
1
B≈0\text{,}116 \;\mathrm{W.K^{-1}}
.
Par contre, pour prendre en compte ce flux il faut connaître l'évolution temporelle des mesures ; or les étudiants n'ont pas mesuré le temps pendant le mélange...
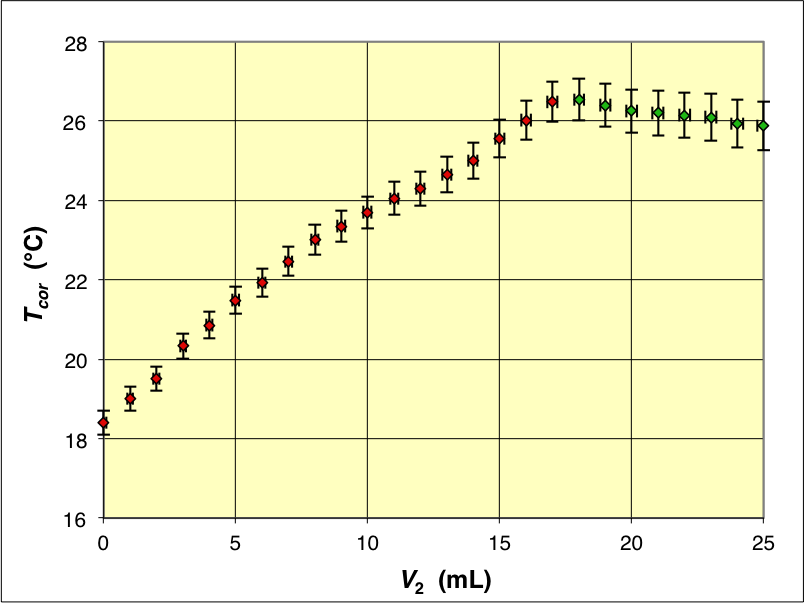
Puisque ces fuites sont faibles, une solution consiste toutefois à supposer que la durée
τ
τ
de chaque mesure reste à peu près la même, puis d'ajuster
τ
τ
pour obtenir
∆
r
H
∆_r H
constant. On obtient ainsi
τ
=
24
±
5
s
τ=24±5\:\mathrm{s}
.
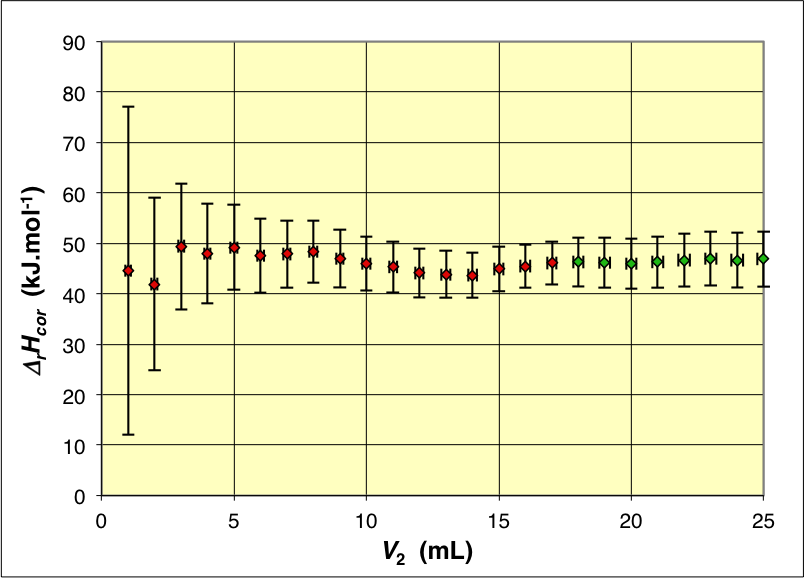
Les variations de température corrigées donnent
∆
r
H
=
46
,
0
±
1
,
0
k
J
.
m
o
l
−
1
∆_r H=46\text{,}0±1\text{,}0 \;\mathrm{kJ .mol^{-1}}
(valeurs plus uniformes). Hélas cela semble difficilement compatible avec la valeur généralement admise
∆
r
H
=
55
,
8
±
0
,
2
k
J
.
m
o
l
−
1
∆_r H=55\text{,}8±0\text{,}2 \;\mathrm{kJ .mol^{-1}}
(à ces températures).
◊ remarque : une interprétation possible est associée à la préparation de
V
1
V_1
à partir de la bouteille de solution mère ; les étudiants transportent dans des béchers et ont interdiction de remettre le reste dans la bouteille ; ces béchers sont transmis à d'autres groupes et il peut y avoir confusion avec les béchers contenant par exemple de l'eau de fusion ou de condensation des autres manipulations ; ceci conduirait à des mélanges de concentration acide inférieure ; ainsi
𝒞
a
≈
0
,
82
m
o
l
.
L
−
1
𝒞_a≈0\text{,}82 \;\mathrm{mol .L^{-1}}
correspondrait à la valeur obtenue.