• En supposant que le système {calorimètre + solutions} est thermiquement parfaite-ment isolé de l'extérieur, on peut considérer que (globalement) . En supposant qu’il est pseudo-isolé mécaniquement et qu’il ne reçoit pas d’autres travaux (électriques, ou autres), on peut considérer que . En supposant qu’il est immobile, on peut considérer en outre que . Le premier principe se limite donc à : .
• En considérant la somme des contributions des différentes parties du système (à pression constante), on obtient ainsi : où décrit le réchauffement du calorimètre et de la solution dosée, où décrit la réaction acido-basique (tant que l’équivalence n’est pas atteinte) et où décrit le réchauffement de la solution dosante.
◊ remarque : les solutions ont des masses volumiques et des capacités calorifiques massiques pratiquement égales à celles de l’eau.
• Pour l’ensemble de la transformation, en supposant constantes les quantités , et , on obtient : et donc : .
• Cette expression peut se mettre sous la forme : où et sont des coefficients positifs. Ceci correspond à : , c’est-à-dire à une fonction strictement croissante.
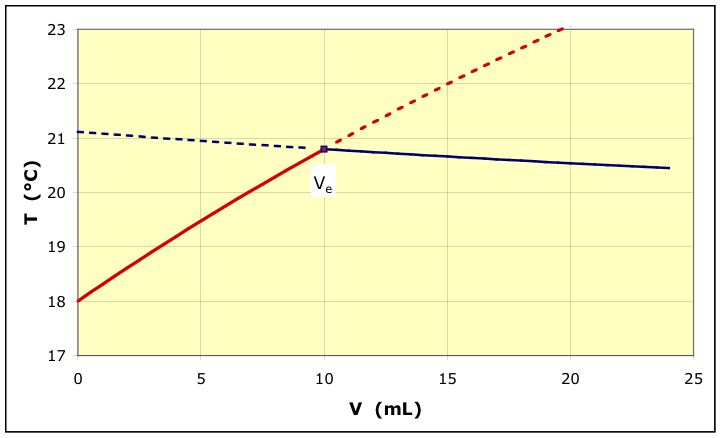
• D’une façon analogue, après l’équivalence : et donc : .
• Cette expression peut se mettre sous la forme : ; donc : , donc la fonction est strictement décroissante. La température maximum est donc effectivement atteinte à l’équivalence : .