at. - ÉTATS DE LA MATIÈRE ET TRANSFORMATIONS
États thermodynamiques ; exemple de l'eau
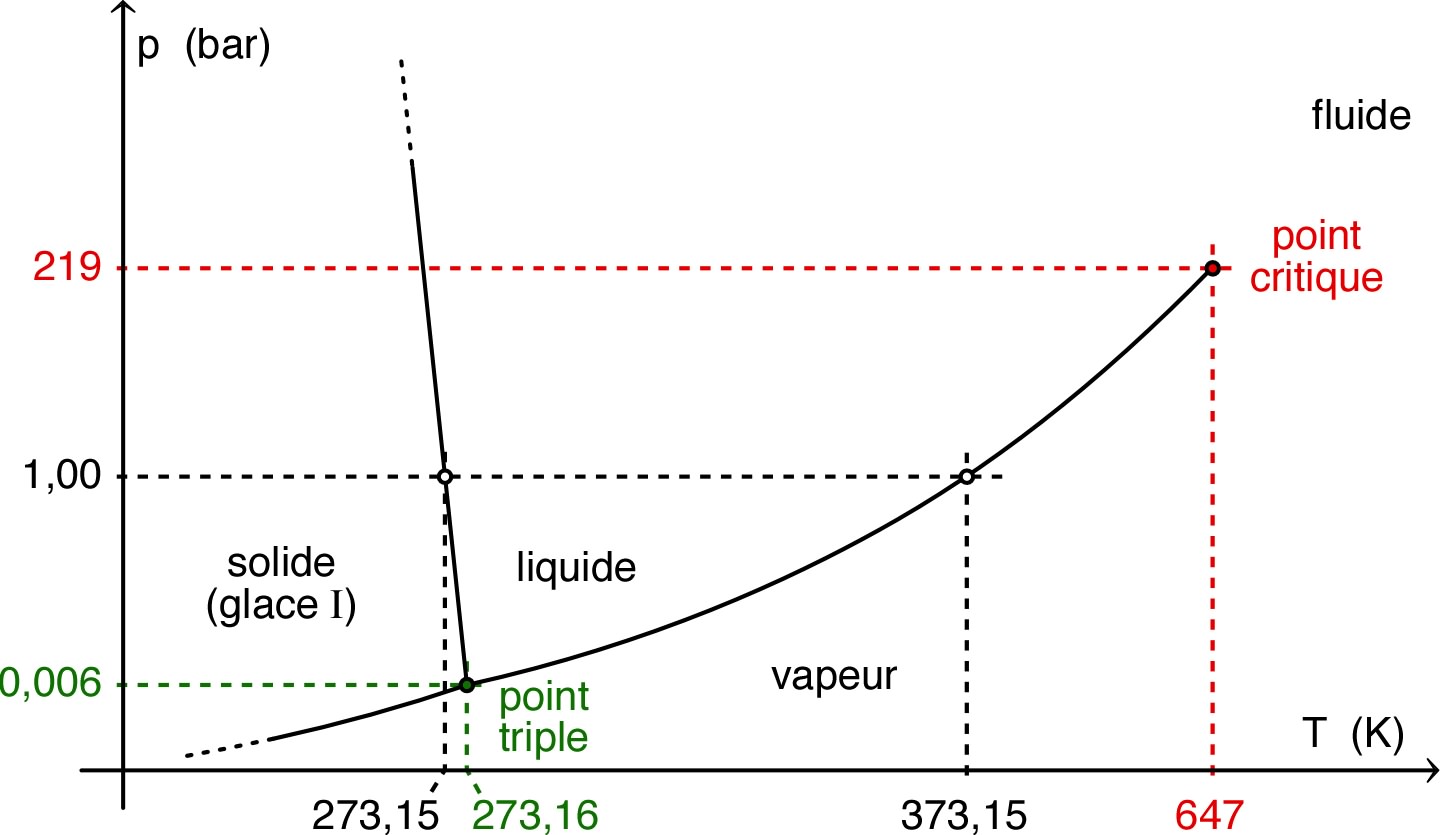
• Le diagramme d'état d'un corps pur, en fonction de la température
et de la
pression
, a une
allure du type représenté ci-après pour l'exemple de l'eau.
On peut y constater la règle de Gibbs sur la
variance :
lors des transformations du système, avec :
(constituants) et
(phases).
Par exemple sur la limite solide/liquide :
(ici
il n'y a que l'eau) ;
(les
deux phases coexistent sur cette courbe) ; ainsi :
(une
seule variable) ; si on fixe la pression cela impose la température
(et inversement).
◊ remarque : le respect des échelles n'est pas possible si on veut
pouvoir distinguer nettement les caractéristiques qualitatives.
• On remarque le point triple (seul où peuvent coexister les trois
phases, ce qui impose
et
) et le point
critique (au delà duquel il n'y a plus qu'une phase fluide, sans
distinction entre liquide et vapeur).
◊ remarque : précédemment, le point triple servait de repère pour
définir l'unité de température, puis on mesurait (par comparaison à
l'unité d'énergie) la constante des gaz parfaits
; depuis mai 2019, l'unité de température se déduit de
celle d'énergie d'après la relation
avec la constante de Boltzmann
et le
nombre d'Avogadro
considérés comme des valeurs exactes (précision
relative
).
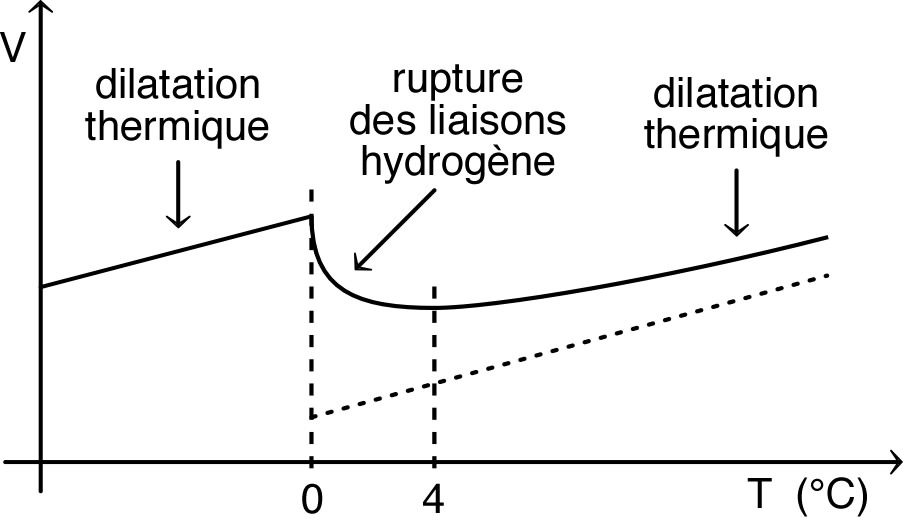
• La structure de la glace (usuelle) étant “lacunaire”, sa masse
volumique est inférieure à celle de l'eau liquide ; c'est pourquoi
la glace flotte sur l'eau.
Plus même : une augmentation de pression favorise la fusion (et non
la solidification) d'où la pente négative de la courbe de
fusion-solidification dans le diagramme d'état (cas particulier
à la cristallisation de l’eau).
Pour une masse donnée, le volume diminue lors de la fusion (par
rupture des liaisons hydrogène), puis ré-augmente (par dilatation
thermique) après un minimum à
.
• D'autres états de la matière existent (selon les espèces et les
conditions) :
- formes cristallisées non uniques (variétés allotropiques) ;
phases vitreuses (liquides “figés“ sans cristalliser) ;
quasi-cristaux (cristallisations moins régulières, selon des
symétries incomplètes) ;
- “cristaux liquides” (structures pseudo-cristallines selon une
ou deux dimensions seulement) ;
- plasmas (gaz ionisés)...
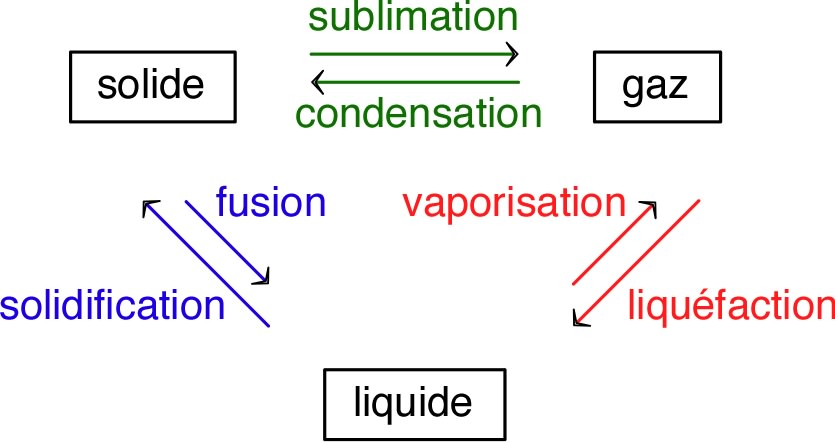
Changements d'état physique
• Les changements d'état physique usuels ont des noms bien définis :
• Il faut ne pas confondre la dissolution dans un liquide avec une
“fusion” (pour un solide) ou une “liquéfaction” (pour un gaz).
L'évaporation est la dissolution d'un liquide (ou d'un solide) dans
un gaz.
◊ remarque : d'autres mots existent dans des cas particuliers
(ébullition, gel...) mais ils peuvent être ambigus ; la brumisation
(formation de fines gouttelettes) n'est pas une vaporisation.
Types de transformations
• À part les transformations d'état physique (par exemple
pour la vaporisation de l'eau), on peut considérer des
réactions :
- nucléaires, par transformation du contenu des noyaux d'atomes
;
- chimiques, par modification des électrons des atomes, ou
modification des groupes d'atomes (liés par interaction de leurs
électrons).
• Les réactions nucléaires les plus usuelles sont les
désintégrations spontanées des noyaux radioactifs :
- désintégration
:
(particule
) ;
- désintégration
:
(antineutrino) ;
- désintégration
:
(positon et neutrino) ;
- “désintégration”
:
(photon).
On peut aussi considérer les réactions provoquées de fusion ou
fission :
- fusion :
(neutron) ;
- fission :
.
• Les réactions chimiques sont de divers types :
- acide-base :
;
- redox :
;
- complexation :
; etc.
• Il est important de savoir reconnaître les différentes sortes de
réaction, tout en sachant qu'elles comportent de nombreuses
analogies.
Avancement de réaction
• Dans un réacteur fermé, les variations des quantités des espèces
chimiques (mesurées en moles) sont proportionnelles aux coefficients
stœchiométriques. Cela peut être repéré par une variable “avancement
de réaction”
(en moles).
Par exemple pour la réaction
:
.
• Pour une réaction écrite sous la forme
générale
:
.
• Ceci permet de décrire l'évolution de la réaction à l'aide d'une
seule variable, avec d'un tableau d'avancement ; par exemple :
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
◊ remarque : on raisonne ici sur le bilan, indépendamment du détail
du mécanisme microscopique (supposé simple pour ne pas influencer le
résultat).
◊ remarque : en réacteur ouvert il faut tenir compte des débits
d’entrée/sortie :
.
• Pour les réactions à volume
constant, on
peut raisonner avec un avancement volumique
(usuellement en
) ; par exemple :
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Activités chimiques
• Pour une espèce chimique donnée, dans des conditions données
(pression, température...), la “tendance à participer à une
réaction” (nommée “activité chimique”) augmente avec sa
concentration.
Pour les solutés relativement dilués (moins de
) l'activité chimique est proportionnelle à la concentration ; on
la définit alors sous la forme (sans unité) :
avec une concentration de référence
.
◊ remarque : dans la mesure où l'usage est de mesurer les
concentrations en
, on écrit souvent par abus de notation
.
| ◊ remarque : pour les espèces plus
concentrées, plusieurs molécules peuvent être en compétition
pour réagir ; l'activité chimique augmente alors moins vite
que la concentration ; par exemple il faut
pour obtenir
. |
|
 |
• Pour les solutions très concentrées,
l'activité chimique est aussi proportionnelle à la concentration,
mais avec un coefficient différent. On choisit alors de définir
l'activité chimique par la fraction molaire :
.
En particulier, dans une solution aqueuse diluée, le solvant
est “presque pur” (
) donc
.
◊ remarque : il en est souvent de même pour les espèces à l'état
solide.
• Pour les espèces à l'état gazeux, dans la
mesure où on raisonne dans l'approximation des gaz parfaits, on
peut exprimer la pression :
(avec
) à partir des “pressions partielles” :
avec
.
On définit alors généralement l'activité
chimique
avec la pression de référence
.
☞ remarque : on peut aussi raisonner en fonction des concentrations,
mais on utilise alors généralement
, ce qui est cohérent avec les pressions en
.
Équilibres réactionnels
Réactions renversables et notion d'équilibre
• En principe, toute réaction qui peut se produire dans un sens peut
aussi se faire dans l'autre sens :
(réaction “renversable”).
Cela peut donner lieu à un équilibre apparent lorsque les deux sens
se compensent :
.
Toutefois, dans certaines circonstances, le “sens inverse” peut être
négligeable ; on dit alors que la réaction est “totale” :
.
◊ remarque : une réaction “totale” est tout de même limitée par
l'épuisement du (des) réactif(s) en défaut, mais l'avancement de
réaction atteint le maximum possible quantitativement.
◊ remarque : pour une réaction renversable, on ne peut pas connaître
directement les avancements respectifs (
et ) des deux
sens, on raisonne en fonction de l'avancement apparent résultant
(algébrique) :
.
Quotient réactionnel et constante d'équilibre
• Pour une réaction écrite sous la forme
générale
on définit le “quotient réactionnel” :
.
Dans le cas d'espèces en solution, on note souvent abusivement
pour simplifier :
;
de même en milieu gazeux :
.
• Lors de l'évolution de la réaction, le
quotient réactionnel tend vers une valeur limite nommée “constante
d'équilibre”, caractéristique de la réaction étudiée. Cette
propriété est nommée “loi d'action des masses”, ou relation de
Guldberg et Waage.
ou
.
◊ remarque : ce sont des constantes par rapport aux espèces
chimiques, mais dépendant de la température
fixée.
◊ remarque : pour les réactions très rapides, dont l'équilibre est
atteint quasi-instantanément, on omet souvent la distinction entre
et
.
Exemple d’application chimique
• La molécule
possède un électron “célibataire” sur l’azote et peut de ce fait se
dimériser (partiellement). Sachant qu’à
et à la pression “normale” (
) la densité du mélange gazeux est
on peut calculer le coefficient de dissociation
:
|
|
|
|
(total) |
|
|
|
|
|
En notant la densité
on peut considérer que la réaction ne change pas la masse et
augmente le volume dans la proportion
(à
pression constante) :
et donc :
.
◊ remarque : en première approximation l'air contient
de
et
de
avec
et
.
• On peut ensuite calculer la constante d’équilibre :
et
;
(
en
évite d'écrire
).
• Si on comprime le système à la pression
à température constante, le coefficient de dissociation
devient :
.
◊ remarque : l’augmentation de la pression déplace l’équilibre dans
le sens qui diminue le nombre de molécules (c'est une propriété
générale).
• Si au contraire, partant de l’équilibre initial, on met le
récipient en communication avec un autre de même volume
, contenant
à
et
, le tout maintenu à volume constant (
) et non
plus à pression constante ; alors il y a dans le mélange
moles de
qui ne participent pas à la réaction, mais déplacent l’équilibre en
modifiant les pressions partielles :
|
|
|
|
|
|
(total) |
|
|
|
|
|
|
|
Ainsi :
;
;
; par suite :
d’où :
et
.
◊ remarque : la dilution déplace l’équilibre dans le sens qui
augmente le nombre de molécules (c'est une propriété générale).
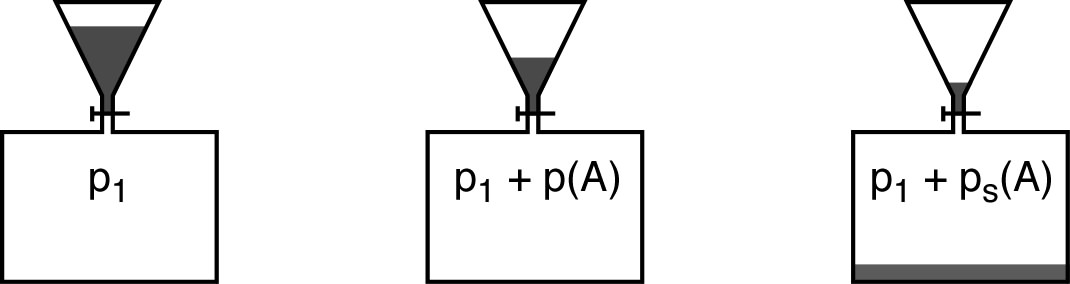
Vapeur “sèche” et vapeur “saturante”
• Lorsqu’on introduit un liquide volatil
dans un
récipient où la pression n'est pas trop grande, l’évaporation fait
augmenter la pression, mais il existe une pression partielle maximum
à laquelle s’établit un équilibre :
:
Compte tenu de la dégénérescence pour le liquide ajouté (pur), la
constante d’équilibre est :
où
est la pression partielle de
.
La pression limite
est nommée “pression de vapeur saturante” ; par opposition, on
appelle “pression de vapeur sèche” la pression partielle de
quand
il n’y a pas saturation.
Par exemple pour l’eau :
;
; .
◊ remarque : pour l'évaporation (ou la vaporisation), la pression
saturante joue un rôle équivalent à celui de la constante de
solubilité d'un solide dans un liquide ; par contre la
fusion
diffère de la dissolution
.
◊ remarque : cette vaporisation se produit quelle que soit la
pression préalable du gaz ; elle est très rapide dans le vide, mais
elle peut être très lente dans un gaz à forte pression (évaporation)
; par ailleurs, pour un mélange liquide, les pressions partielles
des vapeurs correspondantes sont plus faibles, proportionnellement
aux fractions molaires des constituants du liquide.
📖 exercices n° I, II et III.